Brexit: EMA still clear on Timelines
23/05/2018
Despite the currently discussed "transition period", the European Medicines Agency EMA has emphasised that industry should be well prepared even for a so-called "hard Brexit". This was one of the main statements made in an Industry Stakeholder meeting on Brexit and operation of the centralised procedure for human medicinal products, held on 23 March 2018. The European QP Association EQPA was invited to join this meeting; David Cockburn (member of EQPA's Board of Directors) and Lance Smallshaw (Co-Chair of ECA's Executive Board) attended the meeting personally and Ulrich Kissel (Chairman of the EQPA) via remote. They also reported that EMA was clear on the just "agreed" draft withdrawal agreement between EC and UK (only parts of the withdrawal agreement are agreed and so far only by the respective negotiating teams; this does include the transitional arrangements). This agreement will not prevent U.K. becoming a third country starting from 30th March 2019. Any other arrangements might be finalised in 2019 and therefore will come too late to be considered by the companies and EMA´s strategies. It was very clearly recommended to prepare on the scenario of a "hard Brexit" and initiate regulatory changes now in order to have a compliant dossier by the time of Brexit. What is the so-called transition period? Just before the meeting, EU and U.K. negotiating teams agreed the terms of a transition period. This is a compromise, to allow the U.K. to sign its own trade deals during the transition, keeping full free movement rights for EU citizens who arrive during the period in the U.K., as well as automatically implementing new EU rules (which are made without UK input then). In this period there will be no U.K. participation in EU institutions or EU bodies or any Member State meetings (with some exceptions under certain circumstances). This transition period will end on December 31, 2020. As said above, the agreement (a colour-coded text) is not fully agreed yet. How does EMA see this transition period? In the meeting, the EMA pointed out that (pharmaceutical) industry should not rely on this transition period. Currently, the future economic relationship seems to be comparable to the one with for example South Korea or Canada: 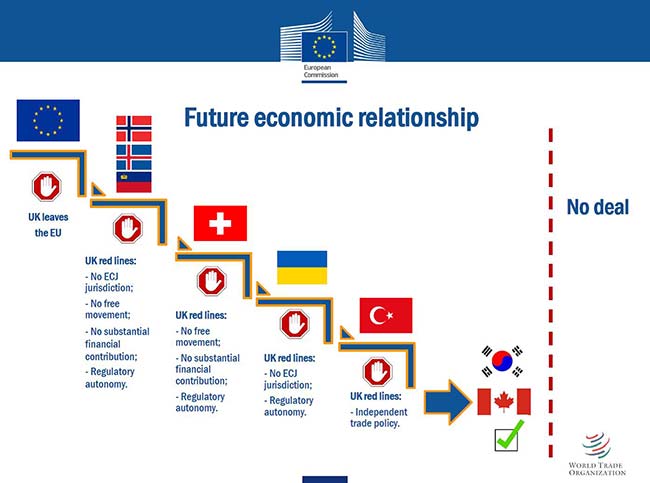
Source: Technical seminar on BREXIT Pharmaceuticals related issues, Brussels, 8 March 2018 What needs to be considered? The EMA pointed out that Marketing Authorisation Holders (MAHs) "need to consider timely process adaptions and changes", like for example: - MAHs need to be established in the EU/EEA;
- Both the QP for batch certification and the QP for Pharmacovigilance (QPPV) need to reside and carry out their tasks in the EU/EEA;
- The site(s) for batch testing and release need to be located in the EU/EEA.
In the meeting most questions concerned regulatory matters like variations and the centralised procedure for human medicinal products and related timeline aspects. However a few discussions were made about operational and GMP aspects: Multi Pack Products: Products that have been placed on the market prior to the date that the U.K. leaves the EU can remain on the market, if the product has been certified by a QP and has already been placed into the distribution chain. But what about multi-packs? Some batches of products released in the U.K. are also used in other Member States with English language (like Ireland, Malta or Cyprus). EMA said that this will not be regulated by EU in future. The agency pointed out that it is not foreseen, that third country labelling information is accepted in the EU. However they leave this as a national regulatory task. EMA also mentioned customs considerations because free movement of goods might not be possible anymore. Acceptance of GMP Certificates: Under current arrangements, after Brexit GMP certificates issued by MHRA can only be used as "supporting information" in risk-based inspection planning. The IWG is currently working on a plan to inspect all third country sites currently relying on a UK GMP certificate so that these hold an EU GMP certificate. The EMA was not clear on how this will affect sites in the U.K. as this will depend on the future arrangements. Safety features: In the meeting, it was said that safety features can be placed on the product by a manufacturing site in the U.K. but the importer (in the EU) is responsible for the correct labelling. In any case, the EU MAH will be responsible for uploading the data to the respective database. (Also see Safety Features for Medicinal Products for Human Use - Questions and Answers Document). Another question was not discussed: serialisation goes live in February 2019 and by 30 March 2019 the U.K. will turn into a 3rd country. So what will be the requirements for these 29 days? This might be a matter for the transition agreement and period. Overall there was a clear message that everyone should prepare now for the fact that U.K. will be a third country after March 2019. Stakeholders should not assume that the draft transitional agreement will extend the time available to make the necessary changes to MAs and batch release arrangements already advised to the industry. The view of the EU Commission In a summary record published after a Technical Expert Seminar on pharmaceuticals related matters with respect to Brexit, the Commission also expressed their concerns about the industry not being ready yet: "Many Marketing Authorisation Holders have not yet taken the necessary regulatory actions, e.g. marketing authorisations transfers, relocation of QPPV etc. While recognising the complexity of some of these procedures for companies to put in motion, there seems to be also a "wait and see" approach by the MAHs. Communication with companies needs therefore to be continued and enhanced reminding them of their responsibility to act swiftly, considering as baseline for preparedness the withdrawal date of 30 March 2019, in order to avoid disruption in the supply of medicines."
Conference Recommendations | |
|
|

