Q&A Session at the QP Forum 2008 in Munich, Germany – Part I
During the Q&A Session at the 3rd QP Forum in Munich, Germany, last December, more than 40 questions asked by the delegates were answered by the speakers team. Following you will find the first set of Q&As:
1. Contract QPs: is there any guidance available defining “sufficient” time on a site to familiarise the QP with the Quality System?
There is no guidance available. My thoughts are that the time on site will depend on the complexity of the quality system. An important consideration is the maturity and stability of the system. If the system is mature and stable a shorter time on site may be indicated, provided that the contract specifies that the QP must be made aware of any changes that affect the quality system.
2. Contract manufacturing: In case of an existing quality agreement, is it sufficient to rely on the certification of other QPs or should there be a review of for example batch records and deviations for the final release?
In theory it is sufficient to rely on the certification of other QPs if the final certifying QP has knowledge of the other QPs and of the quality systems within which each of them is operating. In practice I would recommend that the final QP should be aware of any matter that might affect his/her decision to release (for example deviations or OOS results/investigations) and to review documentation from time-to-time, particularly if contract manufacturers and contract QPs are involved in the process.
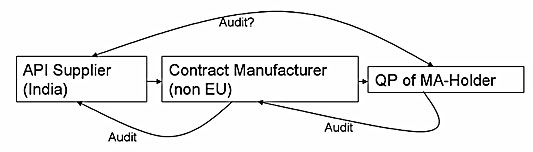
3. How far down the manufacturing supply chain (finished API – intermediate – starting materials) has the QP to place consideration when preparing a GMP API declaration?
Based on a risk-assessment of the process the QP must evaluate the critical materials or critical steps. The QP can base his decisions on statements of authorized persons within a QA-System.
4. How often and with what procedure do authorities expect that in-house specifications and methods are reviewed for being in compliance with the MA?
Based on a controlled Change Control System: every 2 years (during self inspections for example).
5. Is it allowed that a Quality Agreement for a third party audit or audit itself could be performed by the QA of the contract manufacturer (third country), but not QP?
The audit must be performed by a qualified auditor who has no conflict of interest in the company being audited. This would mean that the contract manufacturer could not audit himself, but it would be quite acceptable to have an independent third-party auditor carry out the audit on behalf of the QP. It does not have to be carried out by the QPs themselves.
6. A batch of an API has been released before all testing had been completed. How do I handle this in the release of the final product?
Firstly if this was within the EU then shipment of unapproved API would not be acceptable under EU law. However as this is outside of the EU then the local laws would have to be checked.
It would be necessary to establish what was in the API producers SOP with reference to shipping unapproved material and whether that contravened any regulatory laws. The quality agreement between the API producer, the contract manufacturer and the MA holder should also be looked at to see if there is any reference to the movement of unapproved material being acceptable. It is not good practice to ship unapproved materials and as such this should have been picked up by the Quality person releasing the API and a deviation raised and a full investigation carried out as to the root-cause. In addition the contract manufacturer should have quarantined this material on receipt and also raised a deviation to find out what went wrong. If this was a one-off incident and not a fundamental break down of the API’s manufacturers and/or the contract manufacturers quality system, then as long as the API was formally approved then I would reference the deviation report and as long as everything else was in order, approve the drug product. However, if this incident was part of a systematic failure of the quality system then I would not approve the drug product as the potential for other GMP non-conformances would be to great. The follow-up to the deviation could involve an audit of the API producer initiated by the MA holder.
7. An API is contaminated with very small amounts of glass (<0.02%). The API is micronized and then pressed to tablets (oral). Giving the fact that glass has a very low toxicology; would you release the batch of the final product?
Absolutely not. There is an excellent quote in the European Pharmacopoeia, Chapter 1. “General Notices, Tests and Assays”: “... It is not presumed, for example, that an impurity that is not detectable by means of the prescribed tests is tolerated if common sense and good pharmaceutical practice require that it be absent.”
Good Manufacturing Practice and Good Pharmaceutical Practice require glass particles to be absent in APIs that will be used to manufacture oral solid preparations without any filtration step that would remove the particles!
I assume during its production the API has undergone a last purification step by re-crystallisation after filtration using charcoal or a filter aid. This step should be repeated with the contaminated API (–>reprocessing) to remove the contaminant.
8. A sterility test failed most likely because of a contamination during testing: is a re-test justified?
A retest of a positive sterility test must be very carefully justified based on a root cause investigation giving evidence that there has been a contamination in the laboratory during preparation or testing. It is not appropriate and acceptable to re-test based on mere suspicion.
Reasons to invalidate a positive result would be e.g.
- Microbiological monitoring of the sterility testing facility shows evidence for a failure like detection of the contaminant(s) in the testing environment. This has to be proven by genetic identity of both isolates!
- Microbial growth is found in the negative controls
- After identifying the micro-organisms isolated from the test, the growth of this species can be clearly linked to failures with respect to the material and/or the technique used when conducting the sterility test procedure – e.g. contaminated media or non sterile sterility testing units
9. Raw material for API production: are on-site audits required for all suppliers?
No; however a risk based supplier quality audit programme should be established.
It is important to perform a risk analysis to determine if a supplier needs to be audited. For example in Volume 4 of the EU Guidelines to Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use; Part II Basic Requirements for Active Substances used as Starting Materials it states
7.22 If bulk deliveries are made in non-dedicated tankers, there should be assurance of no cross-contamination from the tanker. Means of providing this assurance could include one or more of the following:
- certificate of cleaning
- testing for trace impurities
- audit of the supplier.
Note that an “Active Substance Starting Material” is a raw material, intermediate, or an active substance that is used in the production of an active substance and that is incorporated as a significant structural fragment into the structure of the active substance.
10. Is a QP responsible to release an API?
Strictly speaking no.
Volume 4 of the EU Guidelines to Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use; Part II Basic Requirements for Active Substances used as Starting Materials does not contain a reference to a Qualified Person.
It is the Quality Unit that has this responsibility for this activity as defined under 2.22 for
- Releasing or rejecting all APIs.
- Releasing or rejecting intermediates for use outside the control of the manufacturing company;
- Establishing a system to release or reject raw materials, intermediates, packaging and labelling materials;
and
2.14 The persons authorised to release intermediates and APIs should be specified.
However the QP is required to certify in an EU marketing application that the API has been manufactured in accordance with Part 2 of EU GMP. It is important to emphasise that it is the QP who is dispositioning the final drug product who has to give the assurance that the API has been made in accordance with the relevant GMPs.
Hence the QP must have access to appropriate documentation including supplier audits (not necessarily by the QP themselves) together with a Quality Contract (Agreement) signed by both parties, to assure themselves that this is the case.
However, member states may have differing national regulations that might require QPs for certain APIs. e.g. in Germany those APIs derived from a human, animal, microbiological source, and manufactured by biotech methods or if the API is used directly as a drug product requiring no additional formulation need a manufacturing licence and a QP.
<< Back to overview
|

